
TL;DR — 台湾の再生医療は「再生医療技術」と「再生医療製剤」の二本の経路に分かれ、二つの法律の中央主管機関はいずれも衛生福利部である。TFDA と医事司は衛福部の部内分業にすぎず、それ自体は法律上の主管機関ではない。日々案件を審査する CDE は二法の中には登場せず、権限は衛福部が《行政手続法》第 16 条に基づいて委託したことに由来する。だから再生医療の一件を処理するには、複数の機関を同時に相手にすることになり、一つの窓口を探せば済むわけではない。この図は、これらの権限の由来と関係を並べて置くことで、どの種類の問題を誰に送るべきかを判断しやすくするものだ。
再生医療の文書が結局誰に送るべきか、どの部署に問うべきかをはっきりさせるために、《再生医療法》と《再生医療製剤条例》を一通り読み、権限関係図を一枚描いた。
AI で図を描くのは難しくない。再生医療技術は《再生医療法》の道、再生医療製剤は《再生医療製剤条例》の道であり、それぞれ条番号を指せる。三つ目の枠が CDE で、その法源はこの二法の中にはない。二法の授権条文には「医薬品査験センター(CDE)」と直接は書かれておらず、書かれているのは「中央主管機関は所属機関に委任し、または他の機関(構)・法人・団体に委託して処理させることができる」である。つまり法律はある種類の単位に委託しうることだけを授権しており、実際に誰に委託するかは《行政手続法》の委託制度と、衛福部が発した委託公告に戻って探すことになる。
そこからこの一篇が答えるべき問いが浮かぶ。日々案件を審査している CDE が演じているのはどのような役割か。法源の根拠は何か。
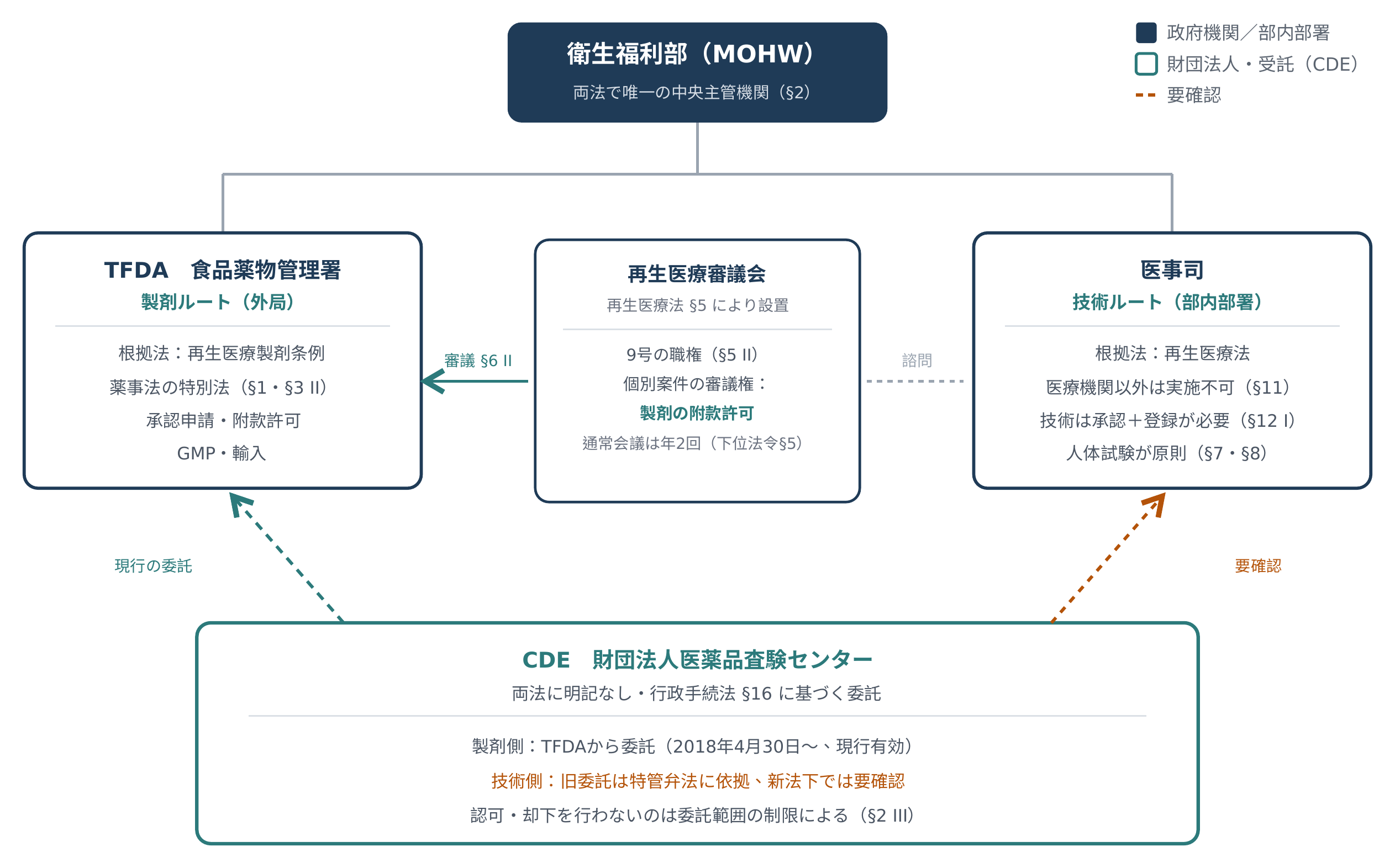
まず完成品をここに置く。以下の六節はこの図の各枠の由来である。技術側の枠は「未確認」とされており、その理由は最後の節にある。
法律上の主管機関は誰か。二法の答えはいずれも衛生福利部だ
多くの人は、TFDA が製剤を、医事司が技術を管轄するから、それらが主管機関だと思っている。だが法律は実際そう書かれていない。
この言い方は機能上は正しいが、法律上はそうではない。《再生医療法》第 2 条と《再生医療製剤条例》第 2 条は、それぞれ同じ一文を書いている。本法(本条例)にいう主管機関は、中央においては衛生福利部とする。
つまり、二法の法定主管機関は同一である。食薬署は衛生福利部の部属機関、医事司は部内の司処であり、いずれも法律上の「中央主管機関」ではない。図の上でこれらを二つの対等なブロックとして並べたなら、描いているのは業務の分業であって、法定の権限ではない。
分業そのものの法理的な基礎ははっきりしている。《再生医療製剤条例》第 1 条後段は「本条例に規定なきものは、薬事法およびその他関連法律の規定による」と書き、第 3 条第 2 項はさらに直截だ。再生医療製剤は「薬事法第六条に規定する医薬品に属する」。一言で再生医療製剤を薬事法体系に釘づけているので、それは自然と食薬署の手に落ちる。《再生医療法》の側は医療側であり、第 11 条は、非医療機関は再生医療を執行してはならないと規定する。
再生医療審議会にはどのような権力があるか。技術法の中に設けられながら、製剤の審議を担う
再生医療審議会の最大の特殊さは、《再生医療法》の中に設けられながら、《再生医療製剤条例》の付款許可を審議することにある。
つまり、それは二法が交わる場所に立っており、「技術」のあの線だけに専属しているのではない。直感的な描き方は医事司のあの線の上に置いて「審議会が承認」と示すことだが、この二点はどちらも間違っている。以下、九款の職権を分解して見れば、どこが間違いかがわかる。
《再生医療法》第 5 条により、審議会は中央主管機関が組織し、条文には「委員若干人を置く」とだけ書かれている(十三から十九人というこの数字は子法《再生医療審議会組織及び運営弁法》第 3 条に由来し、母法にはない)。その職権は第 2 項に九款が列挙され、そのうち七款は純粋な諮問である。すなわち、革新の発展と政策の推進、正しい知識・観念の広報、患者の安全と医療品質の向上、人材育成、再生製剤および再生技術の管理、執行成果の評価、そしてその他の関連事項である。
「審議」の二字を帯びるのは二款のみだ。第五款は「研究開発および奨励・補助の諮問・審議」、第七款は再生製剤の付款許可を交付する審議である。
違いは、第五款が扱うのは補助と奨励であり、第七款が扱うのは製品が付款許可を取得できるか否かという点にある。申請者の前に本当に立ちはだかるあの一款は、第七款だ。
そして第七款は製剤側である。《再生医療製剤条例》第 6 条第 2 項ももう一方から同じことを書いている。中央主管機関が付款許可を交付するには、まず再生医療審議会の審議を経て通過しなければならない。
ここの「しなければならない」は勧告ではなく、法律上の強制手続だ。審議会の審議を経なければ、付款許可を交付できない。
子法はさらに一つ数字を補う。《再生医療審議会組織及び運営弁法》第 5 条はこう書く。本会の会議は年二回の開催を原則とし、必要時には臨時会議を開催できる。
年二回、という意味は、製品が付款許可の道を行くなら、スケジュール表に算入すべきなのは審査日数だけではなく、この委員会がいつ開かれるかもだということだ。一度逃せば原則として半年待つ。同じ条はさらに委員の本人出席を求め、代理の委任を認めず、全委員の過半数が出席しなければ開会できないとする。これらはいずれもスケジュールに影響するが、会議運営の規定に属し、子法の中に書かれていて審査スケジュール表にはないので、自分でめくって探すしかない。
同弁法第 7 条にはもう一つ記憶すべき条がある。審議の準備作業、審議の過程、委員と専門家・学者の氏名は、政府情報公開法に基づいて公開を制限し、会議記録のみを公開する。これは委員が干渉を受けずに審議できるようにするためであり、申請側は名簿を手にできないので、計画時にはまずこの条があることを知っておく必要がある。
医療機関が再生医療技術を執行することについては、承認権は中央主管機関の手にあり、審議会はその線の上では諮問のみである。
付款許可そのものの要件、五年の有効期限と延長不可については、第三篇ですでに一通り分解したので、ここでは繰り返さない。
では CDE は何を根拠にあなたの案件を審査するのか
答えは実のところ《再生医療法》の中ではなく、《行政手続法》の中にある。
CDE は衛生福利部が《行政手続法》第 16 条に基づいて専門審査を委託した法人であり、二法はこれを直接設立していない。二法の中に名を連ねていないことは、法源がないことを意味しない。
第 16 条第 1 項。行政機関は法規に基づき、その権限の一部を民間団体または個人に委託して処理させることができる。
CDE はこの条にいう「民間団体」である。1998 年 7 月に行政院衛生署(衛生福利部の前身)が拠出して設立した財団法人であり、自らの設置条例を持たない。この設計には道理がある。専門審査の内容は科学の進展に伴って変わるので、委託で配置するほうが、そのつど組織法を動かすより柔軟だ。行政手続法第 16 条が与えるのはまさにこの柔軟性である。
第 16 条には三つのキーワードがあり、それらがほぼ CDE の法的位置づけを決めている。「法規に基づき」「権限の一部」そして「委託」だ。
「法規に基づき」は、行政手続法だけでは足りず、個別の作用法の授権を根拠として持たねばならないことを意味する。これらの授権は二法の中に確かに存在する。《再生医療法》第 12 条第 2 項(技術承認)、第 14 条第 3 項(細胞操作の査察許可)、第 18 条第 4 項(細胞保存庫の許可)、そして広告承認のあの二条(再生医療法第 22 条第 2 項、製剤条例第 15 条第 2 項)である。
「権限の一部」は、丸ごと委託に出すことはできないことを意味する。
そして最も肝心なのは「委託」というこの動作の主語だ。前の三条を一条ずつ読むと、文型は一字違わない。「中央主管機関は所属機関に委任し、または他の機関(構)・法人・団体に委託して処理させることができる」。広告のあの二条は書き方がやや異なり、「中央主管機関またはその委任・委託した機関(構)もしくは法人」を用いており、委託の存在を前提としていて委託権を授与しているのではないが、論理は同じだ。言い換えれば、法律が授権しているのは衛生福利部が委託しうることであって、CDE を直接授権しているのではない。
これが三つの単位の法的地位における最大の違いでもある。TFDA と医事司が引き受けているのは法律が直接分配した職権であり、CDE が得ているのは衛生福利部が自らの権限の中から一部を割いて委託した授権だ。法律は「法人に委託しうる」と言うだけで、誰に委託するとは言っていない。
なぜ CDE は承認できないのか。財団法人だからではない
多くの人は、CDE が承認できないのは財団法人だからだと思っている。実はそうではない。
《行政手続法》第 2 条第 3 項は規定する。公権力の行使を委託された個人または団体は、委託範囲内において行政機関とみなす。言い換えれば、法律は CDE が公権力を行使することを禁じていない。
CDE が今日承認権を持たないのは、財団法人だからではなく、衛福部が現在委託している範囲が技術審査と諮問までに限られているからだ。範囲は変えられるものであり、法律はそれを阻んでいない。
この区別は実務上たいへん重要だ。CDE に問えば、返ってくるのは意見である。その意見は重みがあり、あなたが数千万を費やして進むかどうかを左右しうるが、行政処分ではない。もし最終的な結果に不服を申し立てるなら、争う相手は処分を行った主管機関であって、意見を出した受託機関ではない。スケジュールを立てるとき、「CDE が肯定的な意見を出した」ことと「主管機関が承認した」ことは別々のものとして扱わねばならない。
委託公告はどう調べるか。製剤側は調べられるが、技術側は見つけられなかった
まず結論から言う。製剤側の委託根拠は見つけやすい。技術側の、二法施行後の新しい公告は、まだ見つけられていない。
行政手続法第 16 条第 2 項は自分で調べられる取っ手を一つ与えている。委託の際には、委託事項および法規の根拠を公告し、政府公報または新聞紙に掲載しなければならない。言い換えれば、衛生福利部が結局 CDE に何を委託して処理させたのかは、原則として調べられる。
製剤側のこの線ははっきりしている。食薬署は 107 年 4 月 30 日より CDE に医薬品臨床試験計画書、査験登録などの技術的資料審査を委託しており、CDE 自身も 2026 年 2 月に、114 年度に食薬署の委託を受けて IND、BSE、NDA の技術資料評価を執行した成果統計を公告し、この委託が今なお動いていることを証明している。そして《再生医療製剤条例》第 3 条第 2 項がすでに再生医療製剤を薬事法第 6 条の医薬品と性格づけているので、この旧委託は自然とそれを包摂する。再生医療製剤は法律上まさに医薬品なので、もともと医薬品の技術審査に向けた委託が、再生医療製剤にも適用される。この枠は埋まっている。
技術側はもう少し手間がかかる。調べられるのは衛部医字第 1081660965A 号(108 年 3 月 7 日)が公告した細胞治療技術関連の須知であり、その中に細胞治療技術審査業務は衛生福利部が主催し、財団法人医薬品査験センターに関連事務を委託すると記載され、その後 111 年 1 月の版まで改正されている。CDE の公式サイトも、自らが衛生福利部の委託を受けて審査業務を行っていると述べている。
問題は、あの委託の法源が特管弁法であり、その特管弁法はすでに二法の施行とともに失効していることだ。
私は別に新法時代の技術側の委託公告を一つ見つけた。衛部医字第 1151662584 号(115 年 3 月 30 日)で、《再生医療技術及び指定製剤管理弁法》に基づいて診療所の再生医療関連の認証業務を委託するものだ。だがその受託対象は医策会であって、CDE ではない。
二法施行後、衛生福利部が再生医療技術審査について改めて CDE に委託を公告したかどうかは、見つけられなかった。見つからないことは存在しないことと等しくなく、公告が私のめくっていない片隅に散らばっている可能性もある。だがこの枠をありのままに空白にしておくほうが、推論で導いた答えで埋めるより誠実だ。
一通の公文書をその法源に対応させるのがなぜこれほど難しいのか
今年、主管機関とやり取りする過程で、私たちは GMP と GTP の問題を送ったところ、それは彼らの主管範囲にないので食薬署に問うようにという回答を得た。同じ回答の中で、臨床試験段階でどの程度に達すべきかについての CDE の意見が引用されていた。
この回答そのものに問題はなく、二つのことにはそれぞれの根拠がある。製剤の製造規範は薬事法体系に属し、もともと医療技術のあの線の上にはないので、食薬署に戻すのは法に沿っている。CDE の意見がここに現れるのは、それが同時に両側に奉仕しており、しかもそれが出すのはもともと意見であって承認・却下ではないからだ。
難しいのは別のところにある。一通の公文書は自らの権限範囲についてだけあなたに答え、それが押し戻したそのことや、それが引用したその意見が、それぞれどの枠に立っているかを、ついでに説明してはくれない。これは誰の手落ちでもなく、公文書はもともとそう書かれるものであり、各単位も自らが主管する部分についてしか話せない。だがこれは、単位をまたいだあの図を、誰も代わりに組み立てる義務はないということを意味する。自分で戻って条文と公告を一条ずつ読むしかない。それがこの図のやっていることだ。
権力がどこから来るかが、あなたが誰に問うべきかを決める
この図が本当に描いているのは、各単位の権力がどこから来るのかであって、誰が誰より重要かではない。
TFDA、医事司の権限は法律の中に書かれており、法改正がなければ変えられない。
CDE の権限は行政委託に由来し、公告を通じて調整できる。
この違いは二つのことに直接影響する。文書を誰に送るべきか、そして受け取った答えが何を意味するかだ。もともと技術的な意見だけがほしかった問題を主管機関に送れば、返ってくるのはたいてい行政手続上の回答だ。逆に、CDE の技術的意見を主管機関の承認とみなせば、最終的にはやはり正式な承認・却下のあの関門を通れない。二法が施行されて半年、CDE の製剤側の位置は薬事法が受けているので、安定している。技術側を本当に確認したいなら、担当部署に直接問うほうが、公開資料から逆に推論するより速い。
私はもともとある文書をどこに送るべきかを知りたかっただけなのに、最後にはかえって権限構造全体を描き直すことになった。
技術側にはまだ一つ空白の枠が残っており、新法時代の委託公告を今のところ見つけられない。推測するより、まず空けておくほうがよい。見つけたら、また補うことにする。

💬 コメント
読み込み中...